

A process validation report is a document which demonstrates evidence that a manufacturing process is capable of consistently delivering quality products. It provides proper documentation of qualification protocols such as equipment qualification, installation qualification, operational qualification, and performance qualification. Process validation reports are generally completed before the routine commercial production for a new formula or within a new facility and when well-established processes have not undergone a formally documented validation. Quality assurance managers in the pharmaceutical manufacturing industry of the United States typically use a process validation report template to ensure compliance with US Food and Drug Administration (FDA) requirements.
Key Determinants in Validating Manufacturing Processes
The FDA-issued Process Validation: General Principles and Practices is the current guidance for the manufacture of human and animal drug and biological products which aligns process validation activities with a product life cycle approach. An FDA investigative engineer shares that one of the major process validation problems encountered during inspections of US manufacturing facilities is the failure to demonstrate confidence in the process through proper documentation of qualification protocols such as:
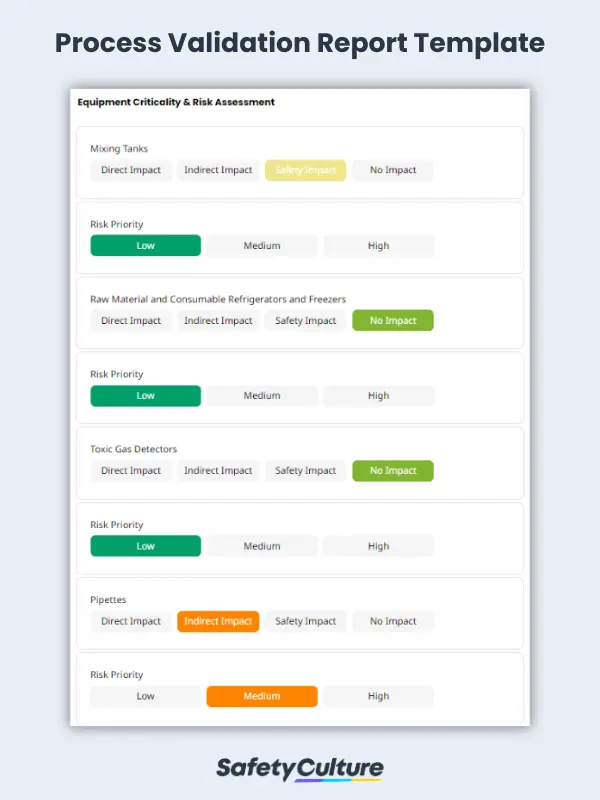
Equipment Qualification
To ensure that all specified design elements have been included and that the design meets the relevant regulatory and statutory requirements, the equipment qualification, also known as design qualification or final design against the user, and functional and/or design specifications should be verified. For adherence to Good Manufacturing Practices (GMP), the equipment at the manufacturing facility should be assessed for criticality according to its designated impact and risk priority.
Installation Qualification
The equipment/system should be installed correctly, supplied as specified, integrated into the manufacturing facility calibration and maintenance systems, and available for use. Supporting utilities should be described and deviations in wiring/cabling, power/fusing, input/output control, etc. should be recorded with a justification for acceptance and impact on operations. The installation qualification protocol should be signed off by the author and approved by the validation supervisor and quality assurance department.
Operational Qualification
The equipment/system should also be operating correctly, compliant with functionality requirements, and integrated into the manufacturing facility training system and Quality Management System (QMS). Calibration equipment, methods, and dates should be included with corresponding Standard Operating Procedures (SOPs); training records; and control point, alarm, and output results.
Performance Qualification
To ensure that the equipment/system is continuously meeting performance criteria for routine use in commercial production, the performance qualification should be verified. For equipment, the normal procedure for each use (configuration or load) should be run three times, and all required data should be recorded. Systems should run for 20 consecutive working days, and deviations to the procedure should be recorded. The acceptance criteria should be compared against the performance test results to formulate conclusions on the validity of the equipment/system.

Jona Tarlengco