
What is GMP Pharmaceuticals?
GMP, meaning Good Manufacturing Practices, is a set of rules and standards that govern how pharmaceutical products are made. It includes specifications for ensuring quality, consistency, and safety in the development, manufacturing, and distribution of medicines. GMP was designed to ensure that safe and effective drugs are produced for patients. It also helps ensure that patients get the right drug at the right dose, with the right side effects, and using the right manufacturing process.
GMP regulations are promulgated by the US Food and Drug Administration (FDA) under the authority of the Federal Food, Drug, and Cosmetic (FD&C) Act. It requires manufacturers, processors, and packagers to ensure that their products cause no harm and are effective.
The International Society for Pharmaceutical Engineering defined GMP pharmaceuticals as “regulations requiring a quality approach to manufacturing, enabling companies to minimize or eliminate instances of contamination, mixups, and errors. This protects the consumer from purchasing a product that is not effective or even dangerous. Failure of firms to comply with GMP regulations can result in very serious consequences including recall, seizure, fines, and jail time.” To avoid such penalties, manufacturers of drugs and medical devices must be able to meet consistent high-quality standards in their production.
Importance of Quality in Pharmaceuticals
Patients or consumers aren’t able to visibly see the quality of drug products. It is mostly assumed that what they will take in their bodies is safe and effective—trusting what was written on the label or packaging. The patient automatically expects quality. They’ll assume that the drug is developed, manufactured, and packaged in a manner that meets industry quality standards and the requirements of regulations such as the FD&C Act. Therefore, it is the duty of the company handling pharmaceutical products to perform quality assurance and quality control at each stage of the process while complying to GMP requirements.
Assurance of Quality
Labels on drug products alone will not give the assurance that consumers and patients expect. According to the World Health Organization (WHO), “Biological products, like any pharmaceutical product, should be manufactured in accordance with the requirements of a Pharmaceutical Quality System (PQS).”
PQS is a quality management system that is used to direct and oversee the processes of a pharmaceutical company in terms of quality. According to the Regulatory Education for Industry (REdI) provided by the US FDA, the real assurance of quality in pharmaceuticals is a complete PQS that includes the following:
- QS elements or framework specified in the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH Q10 on Pharmaceuticals).
- Standards and regulations given by Current Good Manufacturing Practices (cGMP)
- FDA Evaluation by Inspection
Pharmaceutical Quality System (PQS) Elements
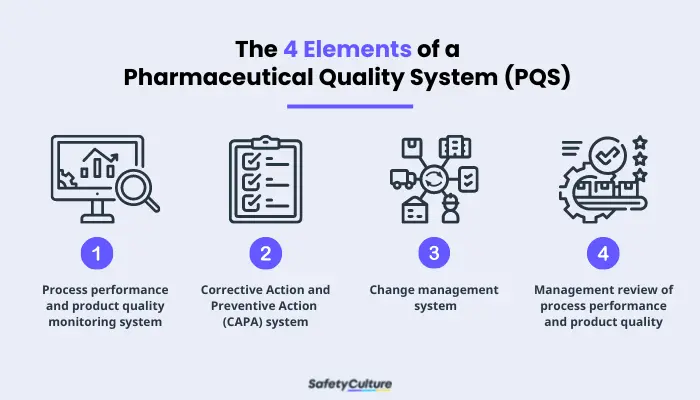
Four Elements of a Pharmaceutical Quality System (PQS)
ICH is the council that brings together “regulatory authorities and pharmaceutical industry to discuss scientific and technical aspects of pharmaceuticals and develop ICH guidelines.” These guidelines are grouped into four: (1) Quality guidelines, (2) Efficacy guidelines, (3) Safety guidelines, and (4) Multidisciplinary guidelines. Q10 guidelines are where ICH focused on PQS and its elements. These elements are:
- Process performance and product quality monitoring system–used to evaluate the performance of processes and identify areas that need to improve.
- Corrective Action and Preventive Action (CAPA) system–used to evaluate the effectiveness of an action.
- Change management system–used in assurance that proper scientific and risk-based assessments are provided.
- Management review of process performance and product quality–used to support continuous improvement.
These four elements of PQS are to be used in ensuring the quality of pharmaceutical products throughout the product life cycle stages.
Achieve operational excellence
Cultivate a culture of excellence with our digital solutions that enhance efficiency, agility, and continuous improvement across all operations.
Explore nowcGMP in Pharmaceutical Industry
The second assurance of quality is cGMP. While GMP and cGMP are mostly used interchangeably, the addition of the term “current” to cGMP intends to remind manufacturers that the system and technology they’re using must be up-to-date, aligned to current standards, or compliant with the latest regulations enforced by FDA.
Manufacturers can avoid most of the common quality problems that put patients at risk, like drug contamination and mix-ups, when they follow cGMP regulations. These regulations set the minimum standards for the procedures, facilities, and controls used to make, process, and package a drug.
Create your own GMP Audit checklist
Build from scratch or choose from our collection of free, ready-to-download, and customizable templates.
Browse GMP Audit checklistsFDA Evaluation by Inspection
FDA is the one who assures the public of a product’s quality and efficacy worldwide. They determine whether or not an organization complies through inspection and evaluation of their product and facilities. FDA does the evaluation through the following:
- FDA’s Compliance Programs–this provides instructions to FDA personnel for conducting activities to evaluate the industry in their compliance with the Federal Food, Drug, and Cosmetic Act.
- Quality System (QS) Regulations–a system consisting of the quality system, the control unit, and all of the validation and review responsibilities associated with quality management.
- 21 CFR 211 (cGMP for Finished Pharmaceuticals)–this governs the manufacturing and specifications of quality products.
SafetyCulture Content Team


