

What is an EU MDR Checklist?
An EU MDR checklist is used to regulate and document the usage of medical devices based on the European Union’s Medical Device Regulations (EU MDR). With a dedicated EU MDR checklist, you can ensure that your medical devices are fit for use and distribution from manufacturing to post-market activities.
Importance
Following the EU MDR requires many documents, inspections, and systems, as it deals with medical devices that will likely be used for life treatment and life-saving. To ensure compliance with all requirements, having a checklist for EU MDR is essential so all necessary steps and documents are followed. It lists down all the tasks you need to fulfill during your medical devices’ production and distribution processes and helps you track and report issues that may arise.
Additionally, by outlining the requirements and procedures that need to be done to comply with EU MDR standards, you can also ensure safety in the workplace. Specifically, you can use your checklist to assist your manufacturers and handlers in identifying potential risks associated with their medical devices. This proactive approach helps in implementing measures to mitigate risks, enhance overall product safety, and improve existing processes.
The checklist also serves as a foundation for creating and organizing necessary documentation. It outlines the documentation requirements for technical files, clinical evaluations, post-market surveillance, and other crucial aspects, ensuring that manufacturers maintain comprehensive records.
What to Include in an EU MDR Checklist
Different devices have different purposes, along with different guidelines to follow under the EU MDR. However, it would be best to have a checklist you can use regardless of your product alongside one specific to your needs.
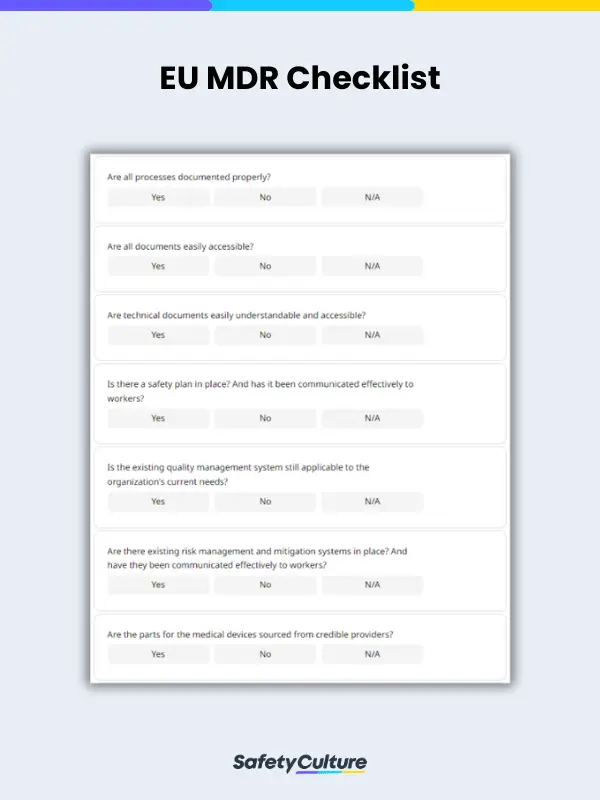
Your general-use EU MDR checklist should have fields for the following:
- Date when the audit is done
- Audit type
- A section with sub-fields for complying with requirements such as:
- Documentation
- Safety plans
- Quality management systems
- Risk management and mitigation plans
- Proper sourcing of materials
- Proper clinical evaluations and trials
- Presence and management of post-market plans
- A section for auditing the important items, parts, and processes involved in the making and distribution of your organization’s medical devices
- Other notes and recommendations
- Inspector’s name and signature
Here is a sample EU MDR checklist in use for reference:

FAQs about EU MDR Checklists
The EU MDR and the US Food and Drug Administration (FDA) are both responsible for regulating the sale and manufacturing of medical devices in their respective territories. Their checklists are similar as well. However, the US FDA also provides regulations for food, medicine, and the like. Meanwhile, the EU MDR is only for regulating medical devices.
There are also differences in their regulations that should be considered in their checklists, such as:
- Classification systems
- Scope of regulations
- Documentation processes required of each organization
All countries part of the European Union follow the EU MDR. Aside from them, other countries that follow this standard include:
- Norway
- Iceland
- Liechtenstein
- Switzerland
There is no specific law requiring the use of an EU MDR checklist. However, to comply with legal regulations and be considered for a Conformité Européenne (CE) certification for distribution in the European region, having a checklist can be very beneficial, as it helps ensure nothing is amiss during your self-audits.

Roselin Manawis